Cells in your organs grow in two distinct ways: they divide to make more cells (called proliferation), and they physically enlarge as they mature. Most organ growth you care about, whether during childhood development or after an injury, comes from cell division driven by a tightly choreographed process called the cell cycle, with mitosis at its center. But here's what surprises most people: cells don't just divide whenever they feel like it. Every step is controlled by molecular signals, checkpoints, and hard physical limits that keep growth useful and prevent it from becoming dangerous.
How Do Cells in Our Organs Grow: Division, Control, Limits

Marcus Whitmore
19 May 2026
The big picture: organs grow through coordinated cell activity
An organ isn't a bag of identical cells all doing the same thing. It's a structured community of many cell types, each with a specific job, all growing in a coordinated way. When your liver regenerates after injury, or your intestinal lining replaces itself every few days, that's not random. Cells in specific zones receive specific signals, divide at the right time, and then stop dividing once the job is done. Growth at the organ level is really thousands of individual cellular decisions being made in parallel, all responding to the same tissue-wide signals.
There's also a common misconception worth clearing up immediately: organ growth is not just cell division. It involves three overlapping processes: cells multiplying (hyperplasia), cells getting bigger (hypertrophy), and cells maturing into specialized types (differentiation). Your heart muscle grows largely through hypertrophy after birth, meaning existing cells get bigger rather than dividing frequently. Your intestine, by contrast, grows and maintains itself almost entirely through rapid division of stem cells. In many organs, these stem cells can grow and regenerate tissue, raising the question of whether they can stem cells grow organs. Knowing which process dominates in which organ is the key to understanding how that organ grows and repairs itself.
Cell cycle basics: how a cell gets ready to divide
Before a cell can divide, it has to go through a preparation sequence called the cell cycle. Think of it like a project with distinct phases that must be completed in order. The cycle has four main stages: G1 (growth and preparation), S phase (DNA replication), G2 (final checks before division), and M phase (mitosis, the actual division). G1 and G2 are often called gap phases, but don't let the name fool you into thinking nothing happens. These are active stages where the cell is growing, repairing DNA, and checking whether conditions are right to proceed.
The molecular engine running all of this is a set of proteins called cyclin-CDK complexes. Cyclins are proteins whose levels rise and fall at specific points in the cycle, and they activate CDKs (cyclin-dependent kinases), which are enzymes that phosphorylate other proteins to trigger the next stage. Cyclin D partners with CDK4 and CDK6 in G1 to push the cell toward S phase by [phosphorylating a protein called pRb](https://www. nature.
com/articles/nrm3567), which releases a transcription factor called E2F that turns on all the genes needed for DNA replication. Later, cyclin B partners with CDK1 to drive entry into mitosis. You can picture cyclins as keys and CDKs as locks: the right key at the right time opens the next door in the sequence.
The restriction point: the cell's point of no return
Late in G1, the cell passes a critical decision point called the restriction point (sometimes called Start in yeast). Before this point, if growth factors are withdrawn, the cell exits the cycle and enters a resting state called G0. After the restriction point, the cell is committed to dividing regardless of external signals. This is why growth factors matter so much early in G1 but not late: the cell is essentially saying, 'I've checked the environment, conditions are good, I'm going ahead.' Understanding this point matters because cancer cells often dismantle this checkpoint entirely, committing to division without waiting for the green light.
Mitosis step by step: making two daughter cells
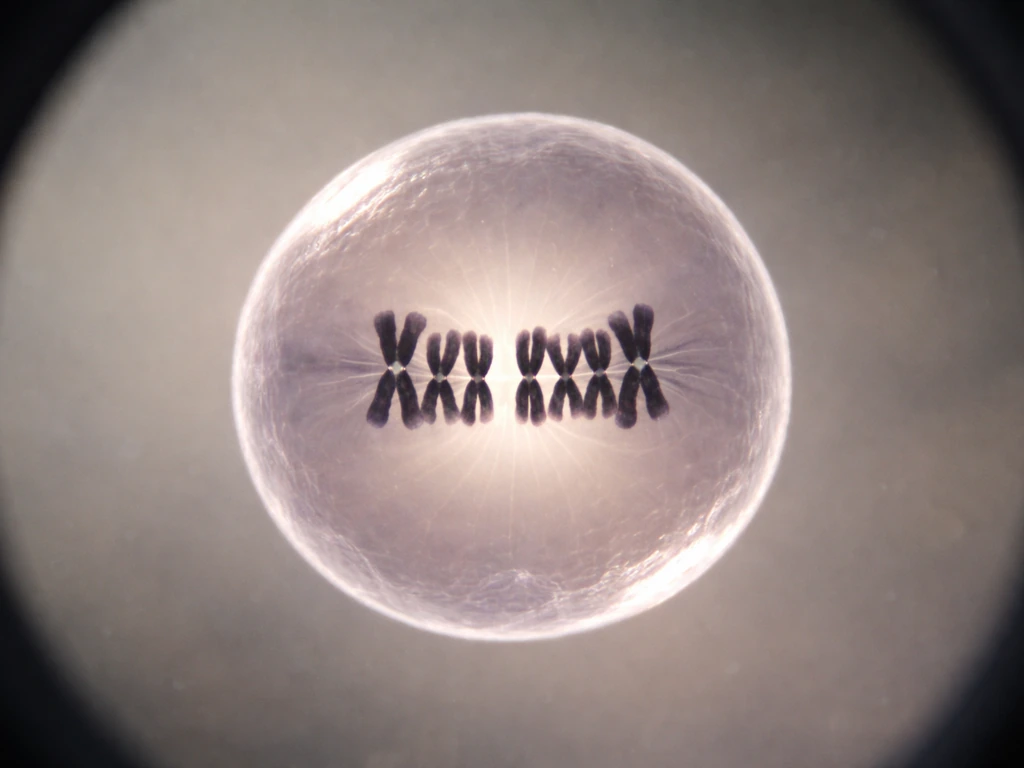
Mitosis is the stage where one cell physically splits into two genetically identical daughter cells. It unfolds through six recognizable stages, and understanding each one helps you see how precise and error-resistant the process is.
- Prophase: The chromosomes condense and become visible under a microscope. The mitotic spindle begins to form from duplicated centrosomes, which start migrating to opposite poles of the cell.
- Prometaphase: The nuclear envelope breaks down, and spindle microtubules reach into the nuclear space to capture chromosomes at structures called kinetochores. Each chromosome must be grabbed from both sides before the cell can proceed.
- Metaphase: All chromosomes line up along the cell's equator (the metaphase plate). This alignment is a quality-control step, not just organization for its own sake.
- Anaphase: The sister chromatids are pulled apart toward opposite poles by shortening kinetochore microtubules and motor proteins. This is the moment chromosomes actually separate, so accuracy here is critical.
- Telophase: Two new nuclear envelopes reassemble around each set of chromosomes, forming two distinct interphase nuclei inside one cell.
- Cytokinesis: The cytoplasm divides, pinching the cell in two. This often overlaps with late telophase and gives you two fully separate daughter cells.
A safety mechanism called the spindle assembly checkpoint (SAC) operates during prometaphase and metaphase to make sure no chromosome is left unattached. The SAC works by inhibiting a protein complex called the APC/C, which is needed to trigger anaphase. Only when every single kinetochore is properly attached to spindle fibers does the SAC release its block, allowing anaphase to proceed. One unattached chromosome is enough to hold up the entire process, which is a remarkable level of quality control.
Signals and control: who tells a cell to grow?
Cells don't decide to divide on their own. They receive external instructions in the form of growth factors, hormones, and signals from neighboring cells. Growth factors are proteins that bind to receptors on a cell's surface and trigger internal signaling cascades that ultimately activate cyclin-CDK complexes and push the cell through G1. EGF (epidermal growth factor), IGF-1 (insulin-like growth factor), and FGF (fibroblast growth factor) are well-known examples, each important in different tissues.
Stem cells play a central role in organ growth because they are the source of new cells throughout life. In the intestine, for example, intestinal stem cells (ISCs) sit at the bottom of small pockets called crypts and divide continuously to produce daughter cells that migrate upward, differentiate, and eventually replace the entire epithelial lining every few days.
Intestinal stem cells reside in specialized instructive niches at the crypt base, and niche-cell signaling controls ISC activity, including homeostasis and regeneration niche signals from neighboring Paneth cells and other support cells at the crypt base. This is one of the fastest cell turnover rates in the human body.
The stem cells themselves are kept in check by niche signals from neighboring Paneth cells and other support cells at the crypt base, which regulate whether a stem cell stays as a stem cell or begins to differentiate. Move too far from the niche and the differentiation signal wins.
Mechanical signals matter too, not just chemical ones. Cells sense how stiff or soft their surroundings are through a process called mechanotransduction. Proteins called YAP and TAZ act as molecular sensors of physical forces from the extracellular matrix (ECM), and they can switch a cell's behavior between proliferation and differentiation depending on substrate stiffness. A cell sitting on a stiff matrix tends to proliferate; on a soft matrix, it may differentiate instead. This is one reason that tissue architecture, not just chemistry, guides how cells grow within an organ.
Checkpoints: the cell cycle's safety net
Three major checkpoints monitor the cell cycle for problems. The G1/S checkpoint checks for DNA damage before committing to replication. The G2/M checkpoint checks that DNA replication was complete and accurate before entering mitosis. And the spindle assembly checkpoint (already mentioned) ensures proper chromosome attachment during mitosis. At each checkpoint, sensor proteins detect problems and activate p53, a transcription factor that activates hundreds of target genes. Depending on the severity of the damage, p53 can halt the cycle to allow repair, trigger permanent arrest (senescence), or initiate apoptosis to eliminate the cell entirely. Think of p53 as the organ's quality-control officer: it would rather sacrifice one cell than let a damaged one keep dividing.
Differentiation: how new cells become the right kind of cell
Cell division produces new cells, but those new cells still have to become the right type for their organ. Differentiation is the process by which a generic progenitor cell commits to a specific identity, switching on a specific set of genes and permanently silencing others. In the intestinal crypt, a newly divided progenitor cell migrating away from the stem cell niche receives signals that push it toward becoming an absorptive enterocyte, a mucus-secreting goblet cell, or a hormone-secreting enteroendocrine cell. The further it travels from the niche, the more committed it becomes, until it is a fully differentiated, non-dividing cell doing its job on the villus surface.
This link between proliferation and differentiation is important to understand: they are largely mutually exclusive states. Rapidly dividing cells tend not to be highly differentiated, and highly differentiated cells tend not to divide. That's why the most actively dividing cells in any organ are often the least mature: stem cells and transit-amplifying progenitors. This trade-off is managed by specific transcription factors and epigenetic changes that lock cells into their final identities. Once fully differentiated, most cells can only divide again if they receive unusually strong signals, such as after serious injury.
What actually limits how much organs can grow
Organ growth doesn't go on forever, and there are several independent mechanisms that put the brakes on it. This is why a question like “can we grow organs” depends on controlling the same signals and checkpoints that determine normal organ size. After transplantation, the transplanted organ can still respond to growth signals, but its ability to grow is constrained by integration, immune control, and the same checkpoints that regulate normal tissue growth transplanted organ grows. Understanding these is just as important as understanding what drives growth in the first place.
| Limiting factor | How it works | Where it's most relevant |
|---|---|---|
| Oxygen diffusion | Oxygen can only diffuse about 100–200 micrometers through tissue; cells farther from a blood vessel become hypoxic and stop dividing or die | Solid tumors, developing organs, wound healing |
| Nutrient supply | Glucose and amino acids must reach every cell; dense tissue without vascularization runs out of metabolic fuel | Rapidly growing organs, engineered tissues |
| Contact inhibition | Normal cells stop dividing when they physically touch neighboring cells, mediated by surface receptors | Epithelial and endothelial tissues |
| ECM and space constraints | The extracellular matrix physically limits how many cells can pack into a space; overcrowding triggers cell extrusion or apoptosis | Epithelial layers, crypts, skin |
| Growth factor depletion | Once a tissue reaches its target size, growth factor levels drop, removing the proliferation signal | Liver regeneration, wound healing |
| Feedback inhibition | Organs release signals (like chalones) that inhibit further proliferation of the same cell type once sufficient cell numbers are reached | Skin epidermis, blood cell production |
Contact inhibition deserves special attention because it's one of the clearest examples of cells responding to their physical context. When epithelial cells reach a confluent monolayer and touch each other on all sides, they stop dividing. Remove a patch of cells and the neighboring cells at the wound edge immediately start proliferating again to fill the gap. This 'wound response' is elegant: the tissue monitors its own density and adjusts proliferation accordingly. Cancer cells lose this response, which is one reason they pile up into disordered masses rather than stopping at a single layer.
When growth stops or goes wrong: apoptosis, aging, and cancer
Not all growth stoppage is a failure. Apoptosis, or programmed cell death, is a clean, controlled removal of cells that are no longer needed or that have become dangerous. It's essential during development (it's how fingers form: the cells between the digits die on purpose) and equally essential in adult life, where it balances new cell production to prevent organ overgrowth. When the balance between cell production and apoptosis is maintained, organ size stays stable. Tip that balance in either direction and you get tissue loss (as in neurodegenerative diseases) or tissue accumulation (as in cancer).
Cellular senescence is a different kind of growth arrest. A senescent cell has permanently stopped dividing but hasn't died. It's triggered by things like telomere shortening, oncogene activation, or persistent DNA damage, and it's enforced by the p53/p21 and p16/Rb pathways. Senescence is actually tumor-suppressive in the short term: it stops a damaged cell from becoming cancerous. But over time, senescent cells accumulate in aging tissues and secrete a cocktail of inflammatory signals called the SASP (senescence-associated secretory phenotype), which can disrupt the tissue environment around them and actually promote chronic inflammation and even nearby tumor development. It's a mechanism that's protective early in life but becomes a liability later.
Cancer is what happens when multiple layers of growth control fail simultaneously. A cancer cell typically has mutations that activate proliferative signaling constantly (like a stuck accelerator), disable tumor suppressors like p53 and pRb (cutting the brake lines), and escape apoptosis. The 'hallmarks of cancer' framework captures exactly this: cancer cells sustain their own growth signals, evade growth suppressors, resist cell death, and eventually co-opt the surrounding tissue to support further expansion.
These same growth-control principles are central to debates about whether scientists should be allowed to grow animals in artificial wombs should scientists be allowed to grow animals in artificial wombs. Understanding normal organ growth, especially the checkpoints and feedback mechanisms described above, is the clearest path to understanding what cancer has broken.
How to study this topic effectively (practical next steps)
If you're a student or educator exploring this topic, a few practical approaches will make the mechanisms click faster than re-reading any summary. But the same basic rules and checkpoints would have to be recreated outside the body, which is why the idea of growing a brain in a lab is far more complicated than just letting cells divide.
But questions like whether it is ethical to grow human organs in the lab also come up, and they involve medical, legal, and moral trade-offs is it ethical to grow human organs. In practice, scientists grow enough cells for research by starting from primary tissue or cell lines and expanding them in culture using optimized media, growth factors, and controlled conditions grow enough cells for their research.
Because organ growth depends on cell division, differentiation, and the stopping signals that keep growth safe, scientists are studying how to recreate those conditions in the lab.
- Draw the cell cycle as a clock with cyclin levels rising and falling as hands: G1 cyclins peak early, S-phase cyclins peak during replication, M cyclins peak at mitosis entry. Annotating your own diagram beats memorizing a textbook version.
- Use mitosis slide images or videos (readily available through open-access biology resources) to identify each phase by the chromosome appearance and spindle structure. Being able to call a phase by looking at a cell is more useful than memorizing definitions.
- When thinking about organ growth, always ask: which cell type is actually dividing here? In most adult organs, it's a stem or progenitor cell, not the differentiated cell doing the organ's main job. Tracking down the stem cell compartment for each organ reveals the real engine of its growth.
- Conceptually practice distinguishing hyperplasia (more cells), hypertrophy (bigger cells), and differentiation (different cells). These three processes are often conflated, and keeping them separate is the foundation of understanding any growth scenario.
- Look into how scientists grow and expand cells in culture for research, because cell culture experiments make abstract concepts like contact inhibition and growth factor dependence very concrete. Related topics on how researchers grow enough cells for their experiments are a natural extension of this material.
- Connect organ growth concepts to the broader questions of whether we can grow organs outside the body or whether stem cells can be used to generate whole organs in the lab, since those topics build directly on the cell cycle and differentiation principles covered here.
Common misconceptions to clear up before you go further
- Cell growth and cell division are not the same thing. A cell can grow (increase in size) without dividing, and a cell can divide without growing much if resources are limited.
- Not all organs grow primarily by cell division. The heart, most neurons, and skeletal muscle cells rely heavily on hypertrophy and rarely divide in adults.
- Stem cells are not embryonic cells frozen in time. Most adult organs have their own resident stem cell populations that are active throughout life.
- Checkpoints do not automatically fix problems: they pause the cycle and give repair machinery a chance to work, but if damage is too severe, the outcome is arrest or death, not repair.
- Apoptosis is not cell damage or necrosis. It's a precise, energy-requiring program that leaves no mess and is essential to healthy development and organ maintenance.
FAQ
Do cells in every organ grow the same way, or does it differ by tissue?
It differs. Some tissues rely mostly on cell division from resident stem cells (for example, intestinal lining), while others grow mainly by cell enlargement after birth (for example, much of the heart muscle via hypertrophy). The “dominant mode” also changes after injury depending on which cell populations are activated.
How do cells actually “stop” dividing once growth is finished?
Stopping usually requires active signals that reduce proliferation pathways, not just passive lack of growth factors. Many tissues also use contact and density cues (contact inhibition) and differentiation programs that lock cells into specialized states, which are generally incompatible with high division.
If differentiation and proliferation are mostly mutually exclusive, can a fully differentiated cell ever re-enter the cell cycle?
Yes, but it is uncommon and usually requires unusual conditions and strong pro-growth cues, such as after serious injury. Re-entry typically demands reprogramming of gene expression and overcoming differentiation-associated cell cycle blocks, rather than simply “receiving a signal” for the next division.
What is the role of the G1 restriction point compared with later checkpoints like G2/M?
The restriction point is a commitment decision in late G1. After it is passed, the cell proceeds toward division even if external growth signals later drop. G2/M checks, in contrast, focus on whether replication was completed correctly and whether it is safe to enter mitosis, so failing G2/M typically halts later rather than preventing the early commitment.
Why can one damaged chromosome stall mitosis, and what does the spindle assembly checkpoint prevent?
The spindle assembly checkpoint delays the transition that separates chromosomes until every kinetochore is properly attached. This prevents mis-segregation that would otherwise produce daughter cells with abnormal chromosome content, which can drive dysfunction or cancerous growth.
How do cells sense the stiffness of their environment, and why does it change whether they divide?
Mechanical sensing converts physical cues from the extracellular matrix into changes in signaling pathways, including through YAP/TAZ activity. On stiffer substrates, these pathways often bias toward proliferation, while on softer matrices they tend to favor differentiation programs.
What happens when the DNA damage response activates p53, does it always lead to cell death?
No. p53 can halt the cell cycle for repair, induce long-term arrest (senescence), or trigger apoptosis if damage is too severe. The outcome depends on damage severity and context, so “p53 activation” is a decision hub rather than a single fate.
Are senescent cells always harmless since they stop dividing?
They are initially protective because they prevent a damaged cell from continuing to proliferate. Over time, senescent cells can accumulate and release inflammatory signals (SASP) that alter the tissue environment, contributing to chronic inflammation and potentially supporting nearby tumor development.
How does apoptosis prevent overgrowth, and where does it show up during normal development?
Apoptosis removes cells that are no longer needed or are harmful, restoring balance between production and tissue size. During development, it shapes structures by eliminating specific cell populations, and in adults it helps maintain organ homeostasis alongside proliferation.
Why do cancer cells often look disorganized compared with normal tissue?
Because multiple growth-control layers fail at once, cancer cells can escape density-based stopping cues, keep proliferative signaling active, and avoid death pathways. The result is mass growth without the normal coordination between cell cycle progression and differentiation.
Can organ growth occur without stem cells?
In most organs, long-term maintenance and regeneration heavily involve stem or progenitor populations. Some growth, like hypertrophy, can occur largely by enlarging existing cells without extensive new cell production, but sustained expansion generally requires a cell source that can supply new specialized cells.
After an organ transplant, will the new organ automatically grow to match the patient’s body size?
Not automatically. Transplanted tissue can respond to growth signals, but its growth is constrained by factors like immune interactions, graft integration into the body, and the same intrinsic checkpoints that control normal tissue size and repair.
What practical mistake do people make when thinking about organ growth?
Assuming growth is only cell division. In reality, organ size and repair depend on overlapping processes, including cell enlargement, differentiation into correct cell types, cell death and clearance, and physical plus chemical stopping cues that prevent unsafe overgrowth.
Next Article
How Do Scientists Grow Enough Cells for Research?
Practical guide to scaling cell cultures: key growth factors, workflows, troubleshooting, and meeting cell quantity targ
