Skin cells grow by dividing in the deepest layer of the epidermis, then slowly moving upward while changing shape and function until they flatten into a tough, protective surface and eventually shed. Red blood cell production, which happens in the bone marrow, also depends on cell division but follows different rules than skin cell renewal do red blood cells grow. The whole journey takes roughly 28 days. The engine behind all of it is mitosis, the same cell division process used across almost every tissue in your body, but skin has a particularly organized way of managing it: a dedicated stem cell zone at the bottom, a strict one-way migration path toward the surface, and a set of molecular signals that decide whether a new cell keeps dividing or commits to becoming part of the barrier. Cells grow in number by doing what: mitosis, which is the core cell division process that expands the population in the basal layer.
How Do Skin Cells Grow? Mitosis, Layers, and Healthy Turnover

Where skin cell growth actually happens
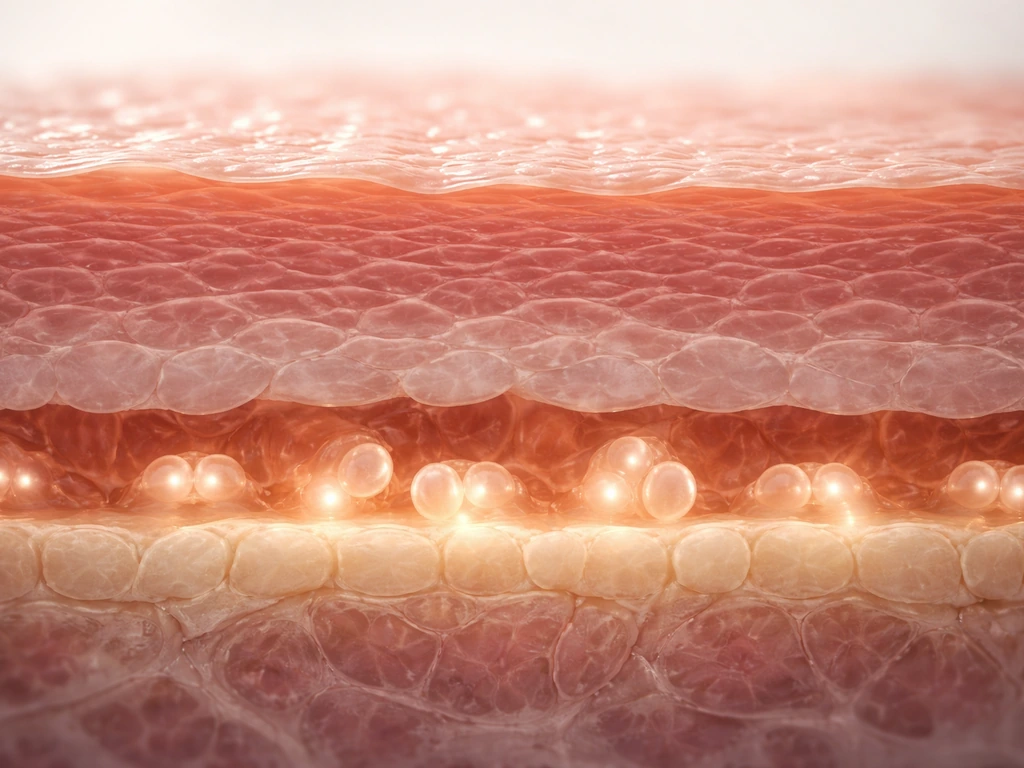
The epidermis is built in layers, and growth only happens in one of them: the stratum basale, or basal layer. Think of it as the production floor of a factory. This single row of cells sits right on top of the basement membrane, a thin protein scaffold that separates the epidermis from the deeper dermis below.
Basal layer cells are called keratinocytes, and the ones with stem-cell-like properties are anchored to the basement membrane through proteins called beta-1 integrins. That physical attachment is not just structural glue: it actively tells the cell to keep proliferating. When a keratinocyte loses contact with the basement membrane, it receives a strong signal to stop dividing and start differentiating instead. Detachment is essentially the starting gun for the long maturation process that turns a dividing cell into a flat, dead-but-useful skin tile.
Above the basal layer are the spinous, granular, and cornified layers (stratum spinosum, granulosum, and corneum). None of these contain dividing cells. They are all the result of cells that were born in the basal layer and have been moving upward ever since.
The cell cycle: what has to happen before a skin cell can split
Before any basal keratinocyte can divide, it has to complete the cell cycle, which is a tightly organized sequence of phases. First the cell grows and copies all of its DNA (S phase), then checks that everything is correct (G2), then actually splits (M phase, when mitosis happens). There are also checkpoint gates built into the cycle that halt progress if something looks wrong, like damaged DNA or missing nutrients.
A key gatekeeper in skin is a protein called p63. It is enriched in basal keratinocytes and acts as a kind of license for continued proliferation. When p63 is active, cells can keep cycling. When p63 is lost or reduced, a cell-cycle inhibitor called p21 ramps up, the brakes go on, and the cell starts down the differentiation path. So p63 is not just allowing division: it is actively suppressing the signals that would push a cell toward maturity too soon.
This matters practically because it shows why skin cell growth is not a simple on/off switch. It is a balance between competing signals, and disrupting that balance in either direction causes problems. Too little proliferation and the skin cannot renew or heal. Too much and you risk uncontrolled growth.
How mitosis actually replaces old skin cells
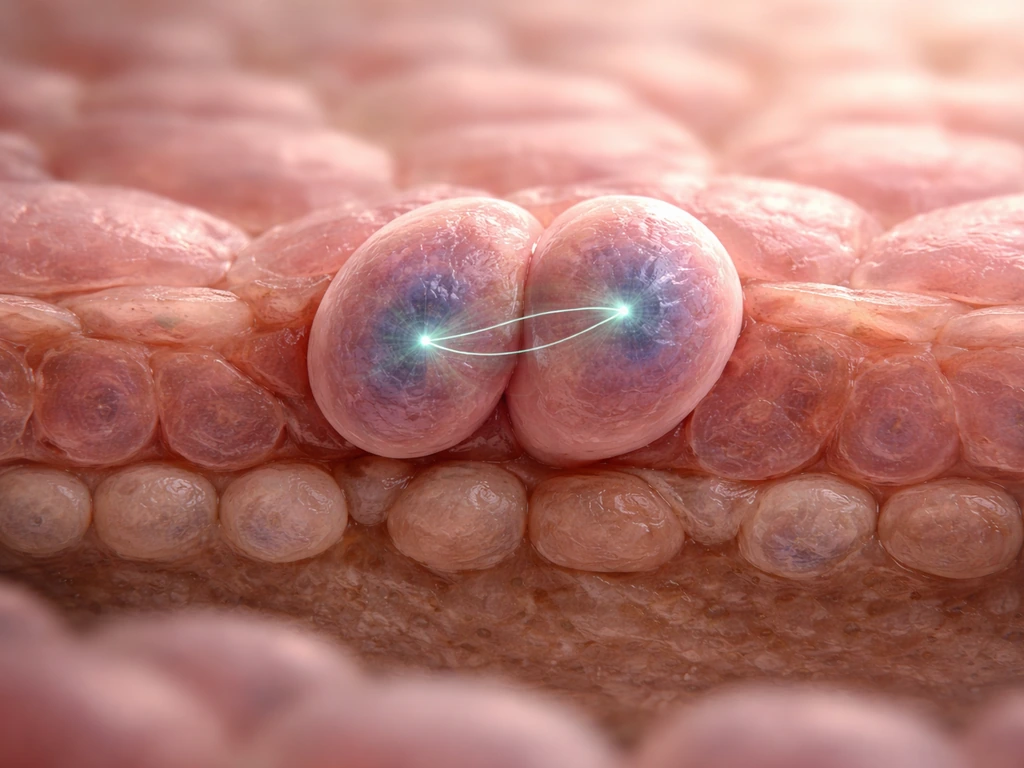
Mitosis in the basal layer is where the numbers get replenished. When a basal keratinocyte divides, the two daughter cells do not have to follow the same fate. This is called asymmetric division, and it is one of the smartest tricks in tissue biology.
The direction the mitotic spindle points during division determines what happens to each daughter cell. If the spindle is oriented parallel to the basement membrane, both daughters land in the basal layer and both retain proliferative capacity: that is symmetric division, which expands the stem cell pool. If the spindle is perpendicular, one daughter stays anchored to the basement membrane (and keeps dividing), while the other is pushed upward, loses that integrin contact, and begins differentiating. The angle of cell division literally determines cell fate.
This is why the basal layer can maintain a stable population of progenitor cells over decades while still continuously supplying the upper layers with new keratinocytes. It is a remarkably efficient system, and understanding it also helps explain why wound healing can ramp up production quickly: the ratio of symmetric to asymmetric divisions can shift depending on the signals present.
Migration, differentiation, and building the barrier
Once a keratinocyte commits to differentiation and moves up from the basal layer, it goes through a predictable series of changes that you can read like a molecular timeline. One of the clearest markers is the keratin switch. Undifferentiated basal cells express keratin proteins KRT5 and KRT14. As soon as a cell exits the cell cycle and enters the suprabasal layers, it switches to KRT1 and KRT10. This swap is not cosmetic: it reflects a fundamental reorganization of the cell's internal skeleton as it prepares for a completely different structural role.
The Notch signaling pathway is one of the main triggers for this transition. When Notch activity increases in a keratinocyte, it pulls the cell out of the cell cycle and kicks off early differentiation markers. It is one of the molecular handoffs that coordinates the shift from a dividing cell to a differentiating one.
As keratinocytes reach the granular layer (stratum granulosum), differentiation becomes terminal. The cells produce large quantities of filaggrin and loricrin, structural proteins that help pack and cross-link the cell's contents into a dense, tough envelope. Loricrin alone makes up about 80 percent of the protein in the cornified envelope by dry weight. Enzymes called transglutaminases cross-link all of these proteins together, creating a structure so sturdy that it essentially replaces the cell's outer membrane. By the time a keratinocyte reaches the stratum corneum, it is no longer really alive in a conventional sense: it is a flattened, protein-filled shell surrounded by a lipid envelope, doing its job as a physical barrier until it sheds from the surface.
The lipid part of the barrier matters too. Lamellar bodies in the granular layer release a mix of ceramides, cholesterol, and fatty acids into the spaces between cells. These form the waterproofing that keeps the skin from losing moisture and keeps irritants out. When this lipid organization is disrupted, as happens in conditions like atopic dermatitis, transepidermal water loss goes up, skin pH rises, and the whole barrier becomes less effective. That is not just a cosmetic issue: it alters the microenvironment that normal cell renewal depends on.
What controls the rate of growth: signals, checkpoints, and limits

Skin cell growth is not just regulated by internal cell-cycle machinery. It is also shaped by physical and chemical signals from the surrounding environment, and several of those signals are specifically designed to prevent overproduction.
One of the most important brakes is contact inhibition. Contact inhibition and confluence-dependent actomyosin contractility suppress proliferation by affecting nuclear localization of beta-catenin and YAP in ligated keratinocytes, as reported in this study confluence-dependent actomyosin contractility suppresses proliferation via beta-catenin and YAP. As keratinocytes crowd together and touch neighboring cells, proteins like E-cadherin engage and trigger a signaling cascade that suppresses proliferation. Part of this works through the Hippo pathway, which inactivates a growth-promoting protein called YAP (Yes-Associated Protein). When cells are sparse or the tissue is damaged, YAP moves into the nucleus and promotes proliferation. When cells are tightly packed and the epidermis is at full thickness, Hippo signaling keeps YAP inactive. You can think of it like a crowded room: once it is full, the door stops letting people in.
Experiments in mouse skin make this vivid. Forcing YAP to stay active in the basal layer causes keratinocytes to keep proliferating well past normal limits, leading to a thickened, abnormal epidermis. Reduce YAP expression and the epidermis gets thinner. The system is genuinely mechano-sensitive: physical crowding, tension, and cell-to-cell contact all feed into the decision of whether to grow or stop.
Calcium concentration is another regulator. Low calcium environments keep keratinocytes in a proliferative state. Higher extracellular calcium, as found in the upper layers of the epidermis, triggers differentiation. This calcium gradient is partly why differentiation happens in a spatial sequence as cells move upward rather than all at once.
On top of all this, oxygen and nutrient supply set a physical ceiling on how fast the basal layer can cycle. Epidermal keratinocytes depend on diffusion from dermal capillaries, and that diffusion distance limits cell density and metabolic rate. It is one of the reasons the epidermis stays thin: going too thick would starve the deeper cells.
Normal renewal vs wound healing: two modes of growth
In normal homeostasis, skin cell growth is calm and steady. If you are wondering how fast do cells grow overall, skin is a good example of how signaling and checkpoints set the pace skin cell growth. Basal cells divide at a rate matched to how fast surface cells shed, keeping epidermal thickness roughly constant. The whole process runs on a roughly 28-day cycle from birth in the basal layer to shedding from the surface, though this varies by body region.
Wound healing is a completely different mode. Within hours of an injury, keratinocytes at the wound edge start migrating toward the gap. Hyperglycaemic conditions have been shown to decrease cultured keratinocyte mobility, consistent with impaired keratinocyte migration relevant to delayed wound healing in diabetes. They do not wait for new cells to be produced first: they physically move, extending lamellipodia and crawling across the wound bed. Growth factor signals, particularly from the inflammatory phase of healing, dramatically upregulate keratinocyte proliferation to generate the supply of cells needed for re-epithelialization.
The wound healing process moves through overlapping phases: an early inflammatory phase handles hemostasis and clears debris, then a proliferative phase brings granulation tissue formation and re-epithelialization, and finally a remodeling phase stabilizes the new tissue. Keratinocyte growth is central to the proliferative phase, but it is tightly coordinated with signals from immune cells, fibroblasts, and blood vessel growth. If any of those conversations break down, re-epithelialization stalls.
The key difference between the two modes is regulatory intensity. In normal renewal, checkpoints keep growth slow and matched to loss. In wound healing, those checkpoints are deliberately overridden by injury signals so that growth can accelerate. Once the wound is closed and contact inhibition is restored across the new epithelium, proliferation slows back down.
What actually helps or hurts skin cell growth in real life
Understanding the biology makes it easier to see why certain habits and conditions matter so much. Here is how the main factors map onto the mechanisms above. In Excel, you can use these same “growth” concepts to control how cells expand when you add or wrap text cells grow with text in Excel.
Things that support healthy skin cell growth
- Hydration and barrier support: Filaggrin and the lipid barrier keep moisture in. Anything that helps maintain that barrier (gentle cleansers, moisturizers with ceramides, avoiding excessive washing) keeps the cellular environment stable for normal turnover.
- Adequate nutrition: Keratinocyte division requires amino acids, vitamins (particularly A and C), zinc, and enough calories. Deficiency in any of these slows the cell cycle directly.
- Sun protection: UV radiation damages keratinocyte DNA, forces cell-cycle checkpoints to halt proliferation, and over time depletes basal progenitor populations. Consistent SPF use preserves the stem cell pool.
- Good blood sugar control: In people with diabetes, high glucose environments reduce keratinocyte migration and proliferation, slowing both normal renewal and wound closure. Glycemic control is genuinely a skin biology intervention.
- Not smoking: Smoking introduces reactive oxygen species and causes tissue hypoxia, both of which impair keratinocyte function and delay the proliferative phase of wound healing. This is not just about aesthetics: it is a direct disruption to the cellular machinery described above.
Things that disrupt the process
- Chronic inflammation: Conditions like atopic dermatitis alter the barrier lipid composition, raise skin pH, and disrupt the normal calcium gradient that regulates differentiation. The result is a cycle where poor barrier function drives inflammation and inflammation drives poor barrier function.
- Repeated physical damage: Chronic abrasion forces the wound-healing proliferation mode to stay active longer than it should, which can eventually exhaust progenitor populations or dysregulate the balance between proliferation and differentiation.
- Certain medications and toxins: Retinoids, for instance, directly alter keratin expression and can shift the balance between proliferative and differentiating states, which is why they are clinically useful but require careful dosing.
- Disrupted contact inhibition signals: Anything that persistently activates YAP or suppresses Hippo pathway signaling can tip the balance toward uncontrolled growth. This is part of the mechanism behind squamous cell carcinoma, where normal contact inhibition breaks down.
Putting it all together
Skin cell growth is a continuous, highly regulated relay race. Stem-like progenitor cells in the basal layer divide using mitosis, with spindle orientation determining whether daughters keep dividing or begin the long journey toward becoming the skin barrier. As keratinocytes migrate upward, they swap keratin proteins, respond to calcium gradients and Notch signals, and progressively build the cornified envelope that protects everything underneath. Contact inhibition, the Hippo/YAP system, and physical limits on nutrient diffusion all work together to keep the epidermis from growing beyond what the body needs. After injury, those limits are temporarily released so that the skin can close wounds quickly, then reinstated once coverage is complete.
If you are interested in how this compares to growth in other cell types, animal cells in general follow similar mitosis-based division cycles, but the differentiation and barrier-building steps are specific to stratified epithelium like skin. The rate of cell growth and the factors that speed or slow division across different tissues follow many of the same rules, just with tissue-specific regulators swapped in. Skin is a useful model for understanding growth constraints precisely because it makes all of this so visible: the layers are literally a spatial map of the growth process, from newborn cells at the base to discarded barriers at the surface.
FAQ
Do skin cells grow the same way in hair follicles and on the rest of the skin surface?
Hair follicles contain their own stem cell populations, and they can help drive regrowth after damage. However, the epidermal layer discussed in the article is the main source of everyday turnover, and follicles and sweat gland ducts do not replace the full-layer keratinocyte cycle.
Is the “about 28 days” timeline always the same for everyone and every body area?
The 28-day cycle is an average, regional turnover varies. Thick skin on palms and soles typically renews more slowly than thinner areas, and turnover also changes with age, sun exposure, and overall health.
What happens to the normal growth schedule after an injury, do skin cells wait for the 28-day cycle?
If the epidermis is disrupted, cells can shift toward faster repair by increasing migration and proliferation at the wound edge, even without waiting for a full normal renewal cycle. In other words, wound healing uses an accelerated program that can bypass the usual timing constraints.
If calcium is low, will skin cells always keep dividing no matter what else is happening?
A low-calcium environment keeps keratinocytes more proliferative, but calcium signaling is not the only determinant. Cell adhesion (like integrin contact), Notch activity, and cell-cycle checkpoints also influence when cells exit the cycle and begin differentiation.
Does nutrient and oxygen supply set a strict maximum for skin cell division, or can the body exceed it during repair?
Nutrients and oxygen mainly cap how fast the basal layer can run, because keratinocytes rely on diffusion from dermal capillaries. But in real tissue, inflammatory signaling, mechanical stress, and barrier state can still make growth temporarily higher or lower within that ceiling.
Can contact inhibition counteract the growth boost from wound healing signals?
Yes, but through pathways the article describes rather than one single mechanism. For example, crowding activates contact inhibition and can reduce YAP-driven proliferation, which shifts the balance back toward slower growth even if injury signals are present.
Can keratinocytes divide in the upper layers (spinous or granular) during normal skin renewal?
The basal layer is where mitosis occurs during normal homeostasis, and suprabasal layers are typically non-dividing. If divisions appear higher up, it usually indicates abnormal regulation or disease rather than normal turnover.
What would happen if basal keratinocytes could not attach properly to the basement membrane?
Integrins support proliferation while cells are attached to the basement membrane, and detachment triggers differentiation. If integrin signaling is impaired, basal cells may not sustain the right proliferative state even if they are in the right anatomical layer.
Is contact inhibition mainly controlled by E-cadherin, or are there other brakes that can change proliferation?
E-cadherin and Hippo/YAP are central, but the “brake” network also depends on the broader tissue context, including mechanical tension and available growth factors. So contact inhibition can be altered in multiple ways, not only by one protein.
How can you tell when a basal keratinocyte has started differentiating rather than simply moving upward?
Differentiation is coordinated across multiple steps, but one common early readout is the keratin switch (from KRT5 and KRT14 to KRT1 and KRT10) as cells exit the cycle. The article’s point is that markers reflect the transition, not just the location.
In wound healing, do skin cells start by growing first or by migrating first?
During wound closure, keratinocytes first migrate to cover the defect, then proliferation increases to supply enough cells for sustained re-epithelialization. Once the epithelium is continuous and contact inhibition returns, proliferation typically drops.
If differentiation markers look normal, can barrier problems still occur due to lipid defects?
Barrier function depends on both the protein envelope and the lipid organization. If the lipid “waterproofing” is disrupted, cells may still differentiate, but barrier performance and the microenvironment needed for balanced renewal can deteriorate, slowing effective repair and promoting irritation.
What are the real-world signs when skin cell growth is too slow versus when it is too fast?
Yes, imbalanced growth can lead to both insufficient renewal and excessive, dysregulated proliferation. When too little happens, healing is delayed and the surface barrier weakens, when too much happens it can cause abnormal thickening and compromised tissue organization.
Do other stratified tissues like the mouth or esophagus use the exact same growth rules as skin?
Skin homeostasis is species and region dependent, and the article’s mechanistic themes generalize across stratified epithelia. Still, the specific regulators and timing can differ, so results from one tissue do not always transfer directly to another.
Do Red Blood Cells Grow? How RBCs Form and Mature
See how RBCs form in bone marrow, why mature red blood cells do not grow or divide, and how anemia changes counts.