Uncontrolled cell growth is dangerous because it breaks the fundamental contract that keeps a body running: every cell does its job, then stops when it's told to. When that stopping mechanism fails, you get cancer. The National Cancer Institute defines cancer as a disease in which cells divide uncontrollably and spread into surrounding tissues. That spread is the key danger. It's not just that cells multiply too fast; it's that they refuse every signal telling them to quit, consume resources meant for healthy tissue, invade organs that can't function with squatters inside them, and eventually shut down systems the body can't live without.
Why We Don’t Want Cells to Grow Uncontrolled: Causes and Next Steps

Marcus Whitmore
5 Apr 2026
What 'uncontrolled cell growth' actually means

Normal growth is purposeful. When you cut your finger, nearby skin cells divide to close the wound, then stop. When a child's bones lengthen, growth plates churn out new cells in a tightly regulated rhythm, then go quiet. Growth that is controlled has an on-switch and an off-switch working in sync.
Uncontrolled growth means the off-switch is broken. Cells divide too quickly and, crucially, do not die in a normal way. MedlinePlus puts it plainly: cancer occurs when genetic material in cells becomes changed so that cells divide too quickly and do not die as they should. The result is a mass of cells that ignores every biological stop sign. Normal growth has goals; uncontrolled growth has none, except relentless multiplication.
It's also worth separating this from simple size. Cells don't just continue to grow larger indefinitely even under healthy conditions, because surface-area-to-volume ratios and nutrient diffusion physics impose hard limits on individual cell size. Uncontrolled growth is not about one cell swelling to enormous proportions; it's about a population of cells replicating without restraint. That distinction matters for understanding why the controls below exist.
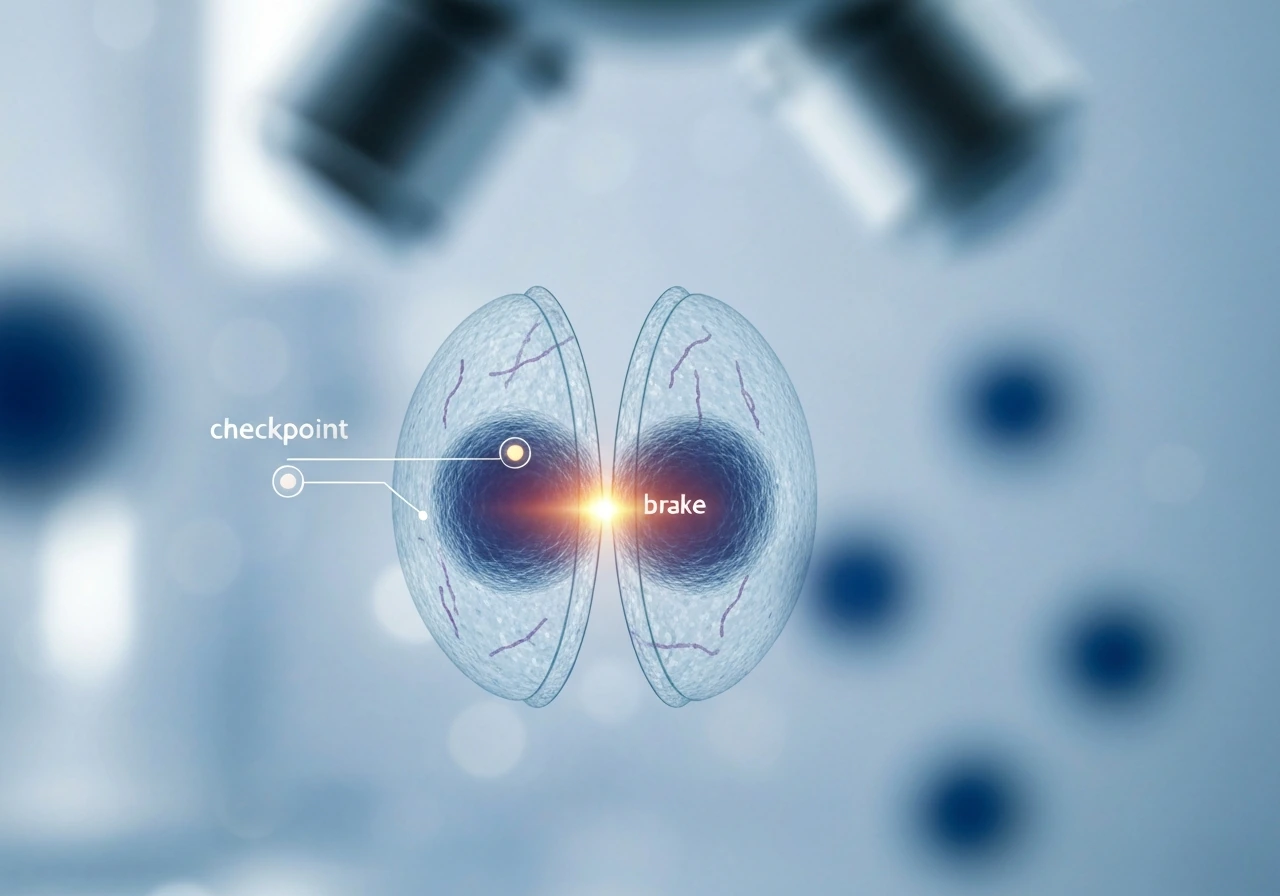
The cell cycle's built-in brakes and checkpoints
Think of the cell cycle as a relay race with referees posted at key handoff points. The referees are called checkpoints, and their job is to ask one question before the race continues: is everything okay to proceed? There are three main checkpoints: G1 (before DNA copying begins), G2 (after DNA copying, before division), and the spindle assembly checkpoint (during division itself).
The G1 checkpoint is the most critical brake. In response to DNA damage, the G1 checkpoint blocks progression into S phase, and p53 can induce p21 to help arrest the cell cycle G1 checkpoint blocks progression into S phase in response to DNA damage. A protein called p53, often described as the "guardian of the genome," scans for DNA damage here. If it finds a problem, it activates p21, which inhibits the cyclin-dependent kinases (CDKs) that would otherwise push the cell into DNA replication. The retinoblastoma protein (Rb) also plays a starring role: when it's dephosphorylated after DNA damage, it clamps down on the genes needed for cell-cycle progression, essentially locking the gate. If damage is too severe to repair, p53 can push the cell toward apoptosis instead of arrest. G1 and G2 checkpoints can both trigger this p53-dependent arrest pathway, meaning the cell has multiple chances to catch a problem before it replicates damaged DNA.
Upstream of p53, signaling proteins called ATM and ATR act like first responders to DNA breaks. They detect damage and relay the alarm down to Cdc25 phosphatases, inhibiting them so that CDK activity stays low and the cycle stays paused. This ATM/ATR pathway is one reason cancer researchers care so much about DNA repair biology. When these alarm systems break down, damaged cells sail through checkpoints undetected.
Signals that tell cells when to grow and when to stop

Cells don't decide to divide on their own. They wait for external permission slips called growth factors, proteins secreted by neighboring cells or arriving through the bloodstream. Epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) are classic examples. They bind to receptors on the cell surface and trigger a cascade of internal signals that say: conditions are right, resources are available, divide now.
One of the most important internal relay routes is the PI3K/AKT/mTOR pathway. A tumor suppressor protein called PTEN acts as a governor on this pathway by converting the signaling molecule PIP3 back to PIP2, which damps down AKT activity. When PTEN is lost, which happens frequently in cancer, PI3K/AKT signaling goes into permanent overdrive and cells get a constant "grow" signal with no counterweight.
Two other stop mechanisms matter enormously here. Contact inhibition is the phenomenon where normal cells stop dividing once they physically touch neighboring cells. Crowd the plate and growth halts. Cancer cells have lost this social awareness. blank" rel="noopener noreferrer">Anchorage dependence is the requirement that normal cells must be physically attached to a surface or scaffold (the extracellular matrix) to divide. Suspend them in fluid and they die. Cancer cells lose anchorage dependence too, which is part of why they can break off, travel through the bloodstream, and set up shop elsewhere. Understanding why cells can't grow too large is one piece of this puzzle, but losing contact inhibition and anchorage dependence is what turns a local growth problem into a systemic one.
Tumor suppressors and apoptosis: the 'stop' and 'self-destruct' systems
Tumor suppressors are proteins whose job is to prevent inappropriate cell division or destroy cells that can't be trusted. p53 and Rb are the most famous examples. p53, when activated, can arrest the cell cycle via p21 or, if damage is irreparable, trigger apoptosis, the cell's built-in self-destruct sequence. Rb restricts the expression of genes needed for DNA replication. When either of these proteins is mutated into a non-functional form, the cell loses two of its most powerful brakes.
How apoptosis actually works
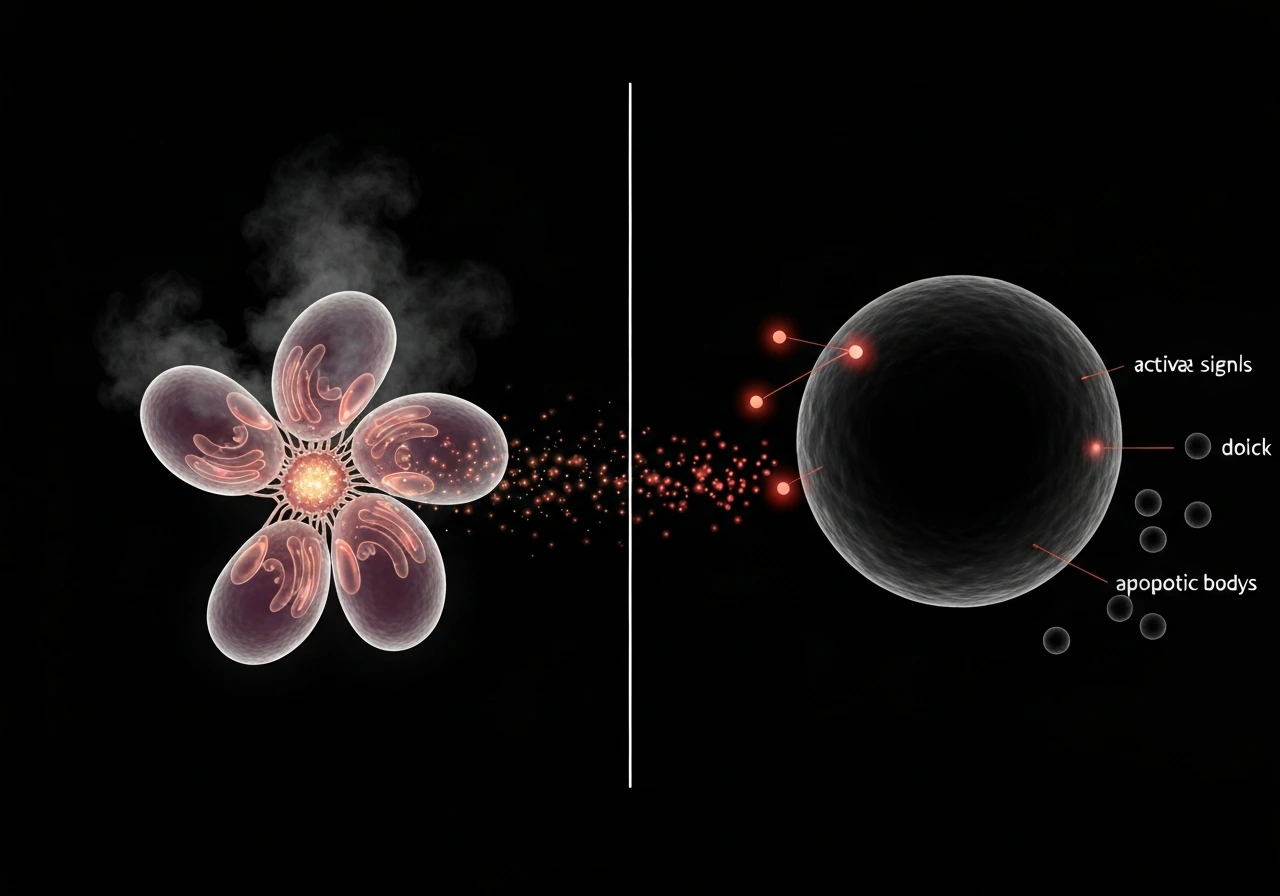
Apoptosis runs through two main pathways. The intrinsic pathway responds to internal stress: DNA damage, oxidative stress, or failed checkpoints. Proteins in the Bcl-2 family, particularly Bax and Bak, trigger mitochondrial outer membrane permeabilization (MOMP). This releases cytochrome c into the cytoplasm, where it joins with a protein called Apaf-1 to assemble the apoptosome, a molecular machine that activates caspase-9 and kicks off a cascade of protein-cleaving enzymes that dismantle the cell from the inside out.
The extrinsic pathway is triggered from outside the cell. Death receptor ligands, molecules like FasL or TNF, bind to death receptors on the cell surface (Fas/CD95, TNFR1). This assembles a death-inducing signaling complex (DISC) that activates caspase-8, which then drives the same execution machinery. Both pathways converge on caspase-3 and caspase-7, the final executioners. Cancer cells frequently block these pathways by overexpressing anti-apoptotic Bcl-2 proteins or by mutating death receptor signaling. They become, in the hallmarks-of-cancer framework, resistant to cell death.
How unchecked growth happens: mutations, broken repairs, and lost regulation
No single mutation causes cancer. It takes a series of hits, usually over years or decades. The first hit might disable one copy of a tumor suppressor. The second might break a DNA repair gene. The third might activate an oncogene, flipping a growth promoter to permanent "on." Each mutation that escapes repair is copied into every daughter cell, building up a genomic disaster incrementally.
DNA repair systems are what normally catch these mutations before they compound. BRCA1 and BRCA2, for example, are critical for repairing double-strand DNA breaks through a precise copying process called homologous recombination. Loss of BRCA function means double-strand breaks go unrepaired or get repaired sloppily, leading to chromosomal rearrangements and genomic instability. Research has documented that inactivation of factors involved in double-strand break repair causes frequent catastrophic genomic events in tumors, accelerating the accumulation of cancer-driving changes.
The hallmarks of cancer framework captures how wide this failure is. Cancer cells don't just divide faster; they sustain their own proliferative signaling, evade growth suppressors, resist cell death, and eventually activate invasion and metastasis. Each of these capabilities corresponds to a broken control system. It's a multi-front collapse of biological governance, not a single malfunction. Cells can't grow indefinitely under normal rules precisely because so many overlapping controls exist. Cancer is what happens when enough of those controls fail simultaneously.
There's also an important category of cells worth knowing: cell cultures that can grow indefinitely are called immortalized cell lines, and they exist precisely because their normal growth-limiting mechanisms have been bypassed, either by viral transformation, deliberate genetic modification, or, in the case of cancer cell lines like HeLa, by the disease itself. These lines are invaluable research tools, but they also illustrate exactly what happens when the brakes come off for real.
Why the body simply cannot tolerate limitless growth
The consequences of unchecked growth go far beyond having too many cells. Here's what actually breaks down:
- Resource depletion: Tumors are metabolically greedy. They commandeer glucose, oxygen, amino acids, and blood supply that healthy tissues need. Tumors cannot grow beyond about 2 mm³ without inducing new blood vessel formation (angiogenesis) to feed themselves, and once they crack that problem, they can grow indefinitely at the expense of the host.
- Loss of tissue organization: Every organ is a precisely arranged community of cell types. A tumor disrupts that architecture, crowding out functional cells and replacing ordered tissue with chaotic growth that can't perform the organ's actual job.
- Invasion and metastasis: Cancer cells that lose anchorage dependence can degrade the extracellular matrix using specialized protrusions called invadopodia, enter blood or lymph vessels (intravasation), travel to distant sites, and colonize new organs. Metastatic disease is responsible for the vast majority of cancer deaths.
- Immune evasion: The immune system normally catches and destroys aberrant cells through a process called cancer immunoediting, which proceeds through three phases: elimination (immune cells destroy early cancer cells), equilibrium (a standoff where some cells persist), and escape (tumor growth outpaces immune control). Cancer cells that survive long enough are precisely those that have evolved ways to hide from or suppress immune surveillance.
- Organism-level failure: When organs can't function, feedback loops that regulate blood pressure, hormone levels, digestion, and respiration collapse. Eventually, uncontrolled growth in one location creates cascading failures that no individual organ can compensate for.
This is also why the question of why we don't want cells growing uncontrolled connects naturally to population biology. Populations of organisms don't grow indefinitely either, for analogous reasons: resource limits, space constraints, and regulatory pressures from the environment impose caps. The biology of limits operates at every scale, from a single cell to an ecosystem. What limits how large a cell can grow is fundamentally a physics-and-chemistry problem; what limits how far a tumor can spread is a biology-and-immunity problem, but both are expressions of the same principle: unconstrained growth destroys the system it's part of.
It's also worth asking: are there any biological entities that grow without these limits? Some things genuinely cannot grow indeterminately for physical and biological reasons, but cancer cells come closer than any other human cell to bypassing those constraints, which is exactly what makes them so dangerous.
Protecting your cells: what the science says you can actually do
Here's where biology meets real life. You can't micromanage your checkpoints, but you can reduce the rate at which your DNA accumulates mutations and make sure that if something slips through, it gets caught early.
Lower your mutation load
Every carcinogen you can avoid is a potential mutation you never have to worry about. Tobacco is the single largest controllable cancer risk: smoking drives lung cancer and contributes to cancers of the mouth, throat, bladder, kidney, and more. Ultraviolet radiation from sun exposure is the primary driver of skin cancers. Alcohol increases risk for liver, esophageal, breast, and colorectal cancers. Maintaining a healthy weight, getting regular physical activity, and eating a diet rich in fiber and vegetables all reduce background inflammation and metabolic stress that can push cells toward instability.
Vaccination against cancer-causing viruses
Some viruses directly damage DNA or hijack growth-control pathways. Human papillomavirus (HPV) drives nearly all cervical cancers and many throat, anal, and penile cancers. The HPV vaccine is recommended for preteens at age 11 or 12, with vaccination available starting at age 9, and the American Cancer Society recommends it between ages 9 and 12 for both boys and girls, with catch-up vaccination through age 26 for those not vaccinated earlier. Hepatitis B infection is a major driver of liver cancer, and the CDC recommends at minimum that all adults get tested for hepatitis B once in their lifetime. These are some of the simplest and most powerful cancer-prevention tools available.
Screening: catching failures before they become disasters
Even with everything above, some cells will accumulate enough mutations over a lifetime to start down the cancer path. That's where screening earns its value. Early detection catches the process when it's still local and still treatable. Here are the current evidence-based recommendations:
| Cancer type | Who | Screening method | Frequency |
|---|---|---|---|
| Colorectal | Adults aged 45–75 | Colonoscopy / annual FIT / stool DNA-FIT | Colonoscopy every 10 yrs; FIT annually; stool DNA-FIT every 1–3 yrs |
| Lung | Adults aged 50–80 with 20+ pack-year smoking history, currently smoking or quit within 15 yrs | Low-dose CT (LDCT) | Annually |
| Breast | Women aged 40–74 | Mammography | Every 2 years (USPSTF guidance as of 2024) |
Talk to your doctor about your personal risk factors. Family history of cancer, known genetic mutations like BRCA1/2, or specific lifestyle exposures may push your screening earlier or more frequently than the population averages above.
What to do right now

- Check whether you're due for any of the screenings in the table above. If you haven't had a colonoscopy and you're over 45, schedule one.
- Confirm your HPV vaccination status (and your children's). If you or a child between 9 and 26 hasn't been vaccinated, talk to a healthcare provider this week.
- Ask your doctor about hepatitis B testing if you've never been tested.
- Identify your top modifiable risk: smoking, sun exposure, alcohol, or obesity. Pick one and make a concrete plan to reduce it.
- Know your family cancer history. First-degree relatives with certain cancers change your risk profile and screening schedule significantly.
The biology behind uncontrolled cell growth is complex, but the takeaway is simple: your body already has extraordinary systems in place to prevent cancer. Checkpoints, tumor suppressors, apoptosis, immune surveillance, and DNA repair all work together to keep division orderly. Your job is to support those systems by reducing mutation-causing exposures, staying current on vaccines, and giving screening a chance to catch any failures early. The cells are doing their part. Meet them halfway.
FAQ
If cancer is just cells dividing too fast, why don’t all fast-growing cells immediately become metastatic?
Tumors still start locally because the first mutations arise in a single clone, checkpoints initially limit spread, and immune surveillance can slow outgrowth. Metastasis becomes likely only after additional changes let cells invade, survive in circulation, and colonize new tissue, which requires multiple failures beyond “fast division.”
Does cancer risk rise with age because cells inherently divide more, or for other reasons?
Age increases risk mainly because time allows more cumulative DNA damage and more opportunities for “hits” to stack up. It is not that older cells have one single switch turned on, rather repair becomes less effective and exposure accumulation (sun, tobacco, infections) increases the mutation load that checkpoints must manage.
Is “uncontrolled growth” the same thing as “uncontrolled cell division,” or can cancer behave differently?
Not always. Some cancers are driven more by evading cell death or sustaining growth signals than by speeding up the cell cycle dramatically. A useful way to think about it is that cancer can be dominated by one broken control (growth, survival, DNA repair, invasion) or a combination, and therapies often target the dominant vulnerability.
How do neighboring cells and the body’s immune system influence uncontrolled tumor growth?
Cancer cells commonly create a permissive microenvironment by recruiting blood vessels, altering immune cells, and changing extracellular matrix so they can invade. This means the “grow” instructions are not purely internal, nearby tissues and signals also help the tumor maintain expansion and escape controls.
Why do different carcinogens lead to cancer in different organs?
Cancer risk varies a lot by exposure type and the organ affected. For example, ultraviolet exposure primarily drives skin mutations, tobacco affects multiple airway and bladder-related pathways through carcinogens, and chronic inflammation can increase reactive oxygen species that damage DNA over time.
How do I know whether screening for my situation is actually worth it?
Early screening is most beneficial when a detectable pre-symptomatic stage exists and when treatment outcomes are better if caught earlier. Screening is not universally helpful for every age group or cancer type, so the right choice depends on your risk profile, test performance, and expected benefit versus harms.
If cancer runs in my family, does that mean I will definitely get it?
You can inherit risk without inheriting cancer itself. Many inherited syndromes (like BRCA1/2) raise the probability by weakening DNA repair or other safeguards, but most cancers still require additional acquired mutations over time, meaning prevention and early detection still matter.
Why can cell lines grow indefinitely in the lab, and should that make me think cancer is inevitable?
Immortalized lab cell lines can be useful because researchers need cells that keep dividing in culture. The important caution is that these cells often have multiple artificial or disease-related changes, so they can behave differently from patient tumors and should not be treated as a perfect “model of you.”
If tumors start locally, why do early cancers sometimes have no symptoms?
Even when cancer starts, symptoms may not appear right away because tumors can grow locally or a person may not notice early changes. This is why screening and risk-based evaluation matter, and also why new or persistent symptoms should be discussed with a clinician rather than waited out.
How do genetic mutations affect both cancer risk and how treatments work?
Some genetic alterations make checkpoint failure more likely, but not everyone responds to the same prevention or treatment approach. Inherited variants can affect repair pathways, and tumor-specific mutations can affect whether cells resist apoptosis or depend on particular growth pathways, which can guide risk assessment and therapy selection.
Next Article
What Controls When and How Fast Cells Grow and Divide
Learn what controls cell division timing and speed: checkpoints, signals, resources, DNA damage, stress, and failure lea
